Investigación FermiNet: Física cuántica y química a partir de principios básicos. En un artículo publicado en Physical Review Research, mostramos cómo el aprendizaje profundo puede ayudar a resolver ecuaciones fundamentales de mecánica cuántica, lo que podría llevar al desarrollo de nuevos materiales y procesos químicos mediante la simulación por computadora antes de su producción. Nuestra red neuronal, FermiNet (Fermionic Neural Network), es adecuada para modelar el estado cuántico de colecciones grandes de electrones, que son los elementos básicos de los enlaces químicos. Esto permite a las comunidades de física y química computacional construir sobre nuestro trabajo y aplicarlo a una amplia gama de problemas.
FermiNet fue la primera demostración del aprendizaje profundo en el cálculo de energía atómica y molecular que fue lo suficientemente precisa como para ser útil, y Psiphor, nuestra nueva arquitectura basada en autoatención, sigue siendo la inteligencia artificial más precisa hasta ahora. Esperamos que las herramientas y ideas desarrolladas en nuestra investigación de inteligencia artificial puedan ayudar a resolver problemas científicos. El enlace covalente sencillo, la base básica de la química, se debe a las interacciones cuánticas de los electrones. Al establecerse las reglas en la década de 1920, los científicos descubrieron que tenían por primera vez una teoría detallada sobre cómo funciona la química. Sin embargo, al intentar calcular realmente las soluciones a estas ecuaciones, se encontraron que solo podían hacerlo precisamente para el átomo más simple (hidrógeno). Todo lo demás era demasiado complicado. Los principios físicos fundamentales necesarios para la teoría matemática de gran parte de la física y toda la química estaban por entonces completamente conocidos, pero su aplicación exacta conllevaba ecuaciones demasiado complejas para ser resueltas. Por lo tanto, se buscó un método práctico aproximado de aplicar la mecánica cuántica. Dirac, fundador de la mecánica cuántica, dijo esto en 1929. A partir de entonces, los físicos desarrollaron técnicas matemáticas que podrían aproximarse al comportamiento cualitativo de los enlaces moleculares y otros fenómenos químicos. Estos métodos se basaban en una descripción aproximada del comportamiento de los electrones, la cual es similar a la química introductoria. Esta descripción asigna a cada electrón un orbital específico y esta asignación depende de la forma media de todos los demás orbitales. La dificultad radica en que esta imagen de Por ejemplo, la energía total de los electrones en una molécula de butadieno es casi 100.000 kilocalorías por molécula, mientras que la diferencia de energía entre las distintas formas posibles de la misma molécula solo es de 1 kilocaloría por molécula. Esto significa que para predecir correctamente su forma natural, se necesita precisión igual a la medida del campo de fútbol hasta el milímetro. Después de la Segunda Guerra Mundial, con la aparición de la computación digital, los científicos desarrollaron una amplia gama de métodos computacionales que pasaban de la descripción media de campos electrónicos.
Estos métodos están llenos de abreviaturas y generalmente caen en algunos lugares en un eje que negocia la precisión con la eficiencia. En uno de los extremos, se encuentran métodos prácticamente exactos que escalan peor que exponencialmente con el número de electrones, mientras que en el otro, se encuentran métodos que escalan linealmente pero no son muy precisos. Estos métodos computacionales han tenido un impacto enorme en la práctica de la química — el Premio Nobel de Química 1998 fue otorgado a los creadores de muchos de estos algoritmos. A pesar de la amplitud de las herramientas mecánicas cuánticas computacionales existentes, se necesita un nuevo método para abordar el problema de la representación eficiente. Hay una razón por la que los cálculos químicos cuánticos más grandes solo pueden ser ejecutados en las decenas de miles de electrones aún con los métodos más aproximados, mientras que las técnicas clásicas como la dinámica molecular pueden manejar millones de átomos. La descripción del estado de un sistema clásico es fácil — solo necesitamos seguir la posición y el impulso de cada partícula. Representar el estado de un sistema cuántico es mucho más complicado — una probabilidad tiene que ser asignada a cada configuración de posiciones de electrones. Función de onda, que asigna un número positivo o negativo a cada configuración de electrones y la función de onda al cuadrado da la probabilidad de encontrar el sistema en esa configuración. El espacio de todas las configuraciones posibles es enorme — si tratamos de representarla como una cuadrícula con 100 puntos a lo largo de cada dimensión, el número de posibles configuraciones de electrones para un átomo de silicio sería mayor que el número de átomos en el universo. En los últimos años, se han hecho enormes avances en la representación de probabilidades complejas y de alta dimensión. Ahora sabemos cómo entrenar estas redes de manera eficiente y escalable. Los investigadores, entre ellos Giuseppe Carleo y Matthias Troyer, han demostrado que el aprendizaje profundo puede ser útil para abordar problemas cuánticos idealizados. En particular, se ha propuesto utilizar redes neuronales profundas para tratar problemas realistas en química y física de la materia condensada. Esto implica incluir electrones en los cálculos, lo que requiere abordar el principio de exclusión de Pauli debido a que los electrones son una clase especial de partículas llamadas fermiones, que tienen una función de onda antisimétrica. Por lo tanto, se desarrolló un tipo nuevo de red neuronal llamado FermiNet, que es antisimétrico en sus entradas.
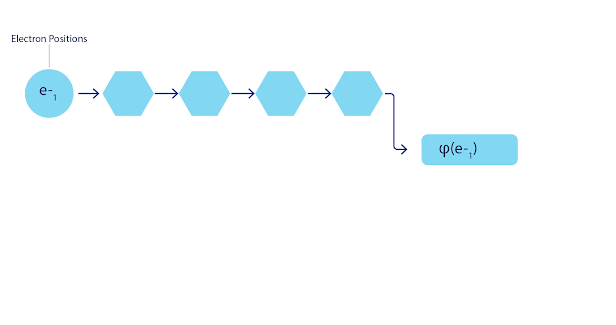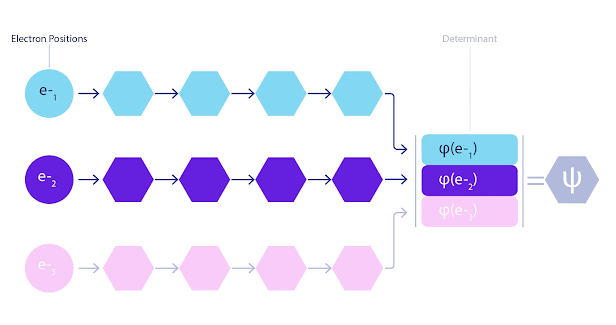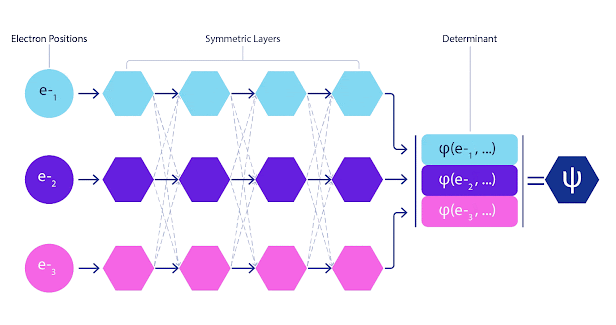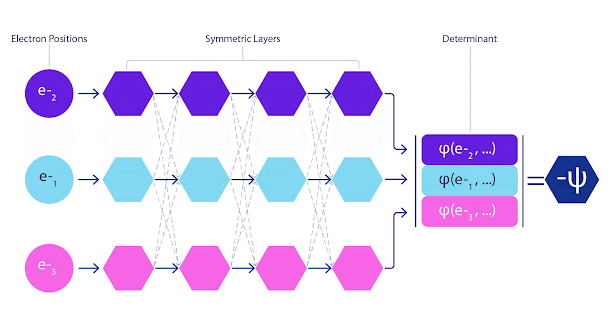
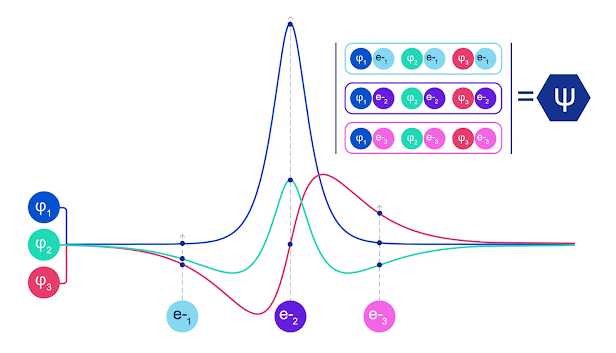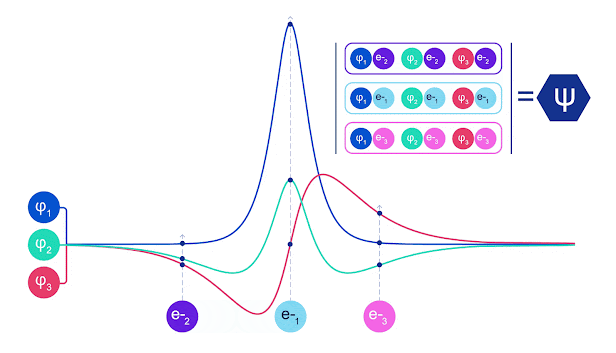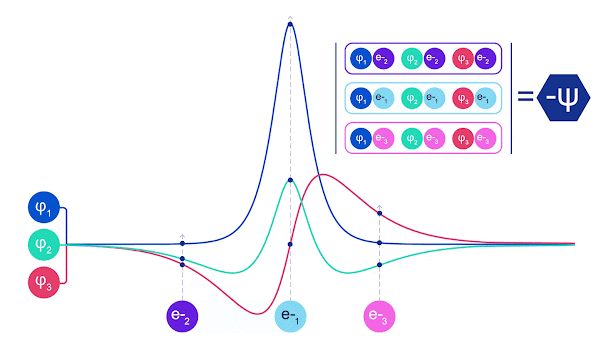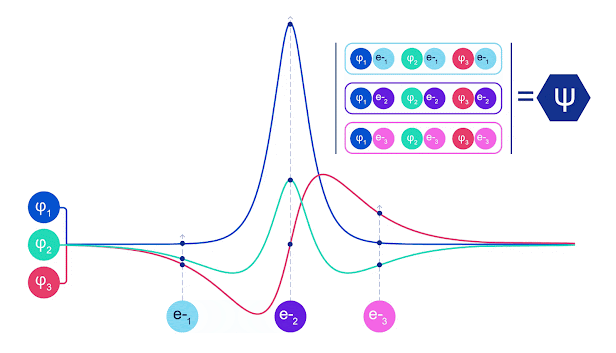
La mayoría de los métodos cuánticos químicos introducen la antisimetría utilizando una función determinante. Las funciones de electrones se evalúan para cada electrón en el sistema y luego se agrupan en una matriz, la cual tiene un factor determinante que es una función de onda realmente antisimétrica. Sin embargo, la principal limitación del enfoque es que la función resultante, conocida como determinante de Slater, no es muy general y los sistemas reales son mucho más complejos. Aun así, esto puede no ser suficiente para calcular las energías. Animación de un determinante Slater. Cada curva representa una sección a través de uno de los orbitales mostrados arriba. Las filas del determinante Slater intercambian posiciones, y la función de onda se multiplica por -1 para garantizar que el Principio de Exclusión de Pauli se cumpla. Las redes neuronales profundas a menudo son mucho más eficientes en representar funciones complejas que combinaciones lineales de bases funcionales. En FermiNet, esto se logra haciendo que cada función que entra en el determinante sea una función de todos los electrones (véase Este va mucho más allá de los métodos que sólo utilizan una y dos funciones de electrón. FermiNet tiene un flujo separado de información para cada electrón. Sin ninguna interacción entre estos flujos, la red no sería más expresiva que un determinante Slater convencional. Para ir más allá de esto, se toma la media de todas las corrientes en cada capa de la red y se le pasa a cada flujo en la siguiente capa. De esta manera, estos flujos tienen las propiedades de simetría correctas para crear una función antisimétrica. Esto es similar a cómo las redes neuronales gráficas agregan información en cada capa. A diferencia de los determinantes Slater, FermiNets son aproximadores universales de la función, al menos cuando se superan las capas de la red neural.
FermiNet presenta interacciones simétricas entre flujos, lo que hace que la función de onda sea mucho más general y expresiva. El determinante convencional de Slater aún cambia las posiciones de electrones conduce a intercambiar filas en el determinante y multiplicar la función de onda global por -1. Minimizamos FermiNet al minimizar la energía del sistema, pero debido a que deberíamos evaluar la función de onda en absoluto para hacerlo exactamente, lo hacemos aproximadamente en su lugar. Elegimos una muestra aleatoria de electrones y calculamos la energía localmente en cada configuración de electrón, sumando las contribuciones de cada configuración y minimizando esto en lugar de la energía verdadera. Este método se conoce como un método de Monte Carlo, ya que es algo así como un jugador de dados rodando una y otra vez. Aunque sea aproximado, siempre podemos volver a tirar los dados para hacerlo más preciso. Debido a que la función de onda al cuadrado da la probabilidad de observar una disposición de partículas en cualquier lugar, es más conveniente para generar muestras propias de la propia función de onda — fundamentalmente, simulando el acto de observar las partículas. Las redes son entrenadas a partir de algunos datos externos; en nuestro caso, las entradas utilizadas para entrenar la red neural son generadas por la red neural en sí. Esto significa que no necesitamos ningún dato de entrenamiento adicional más que las posiciones de los núcleos atómicos que el programa utiliza. La idea básica, conocida como cuántico variable Monte Carlo (VMC), ha estado alrededor desde los años 60 y generalmente se considera una forma económica pero no muy precisa de calcular la energía de un sistema. Las funciones de onda simples basadas en determinantes Slater con FermiNet, hemos aumentado dramáticamente la precisión de este enfoque en todos los sistemas que hemos visto. Los electrones simulados de FermiNet se mueven alrededor de la molécula de biciclobutano. Para garantizar que FermiNet representa un avance en el estado del arte, comenzamos por investigar sistemas sencillos y bien estudiados como los átomos en la primera fila de la tabla periódica (hidrógeno a través del neón). FermiNet supera los cálculos comparables de VMC por un amplio margen, usualmente reduciendo el error relativo en los cálculos escalonales exponenciales hasta un grado significativo. En sistemas más grandes, los métodos de escalamiento exponencial se vuelven inviables, por lo que en su lugar utilizamos el método del cluster acoplado como base.
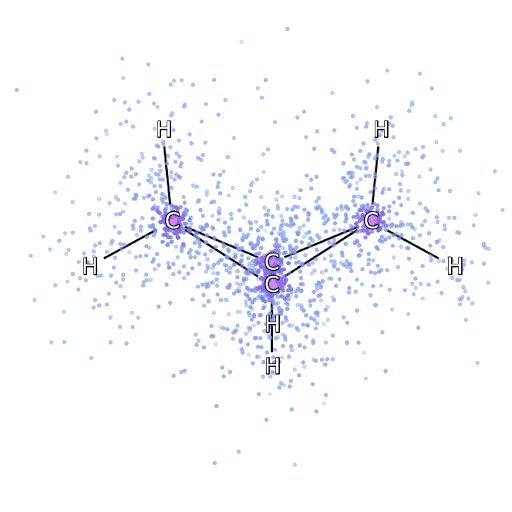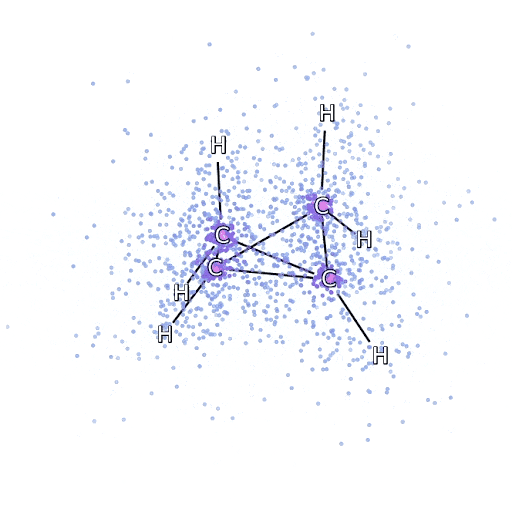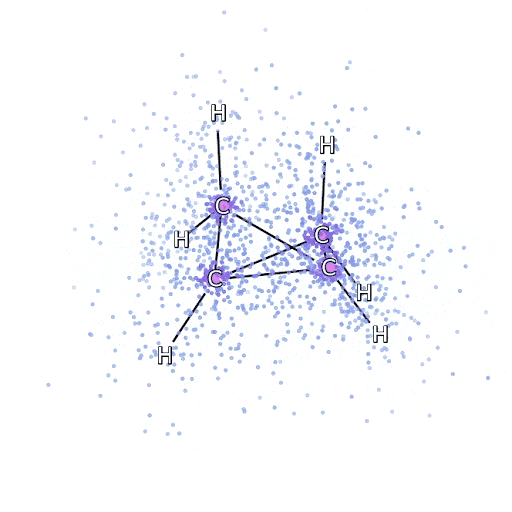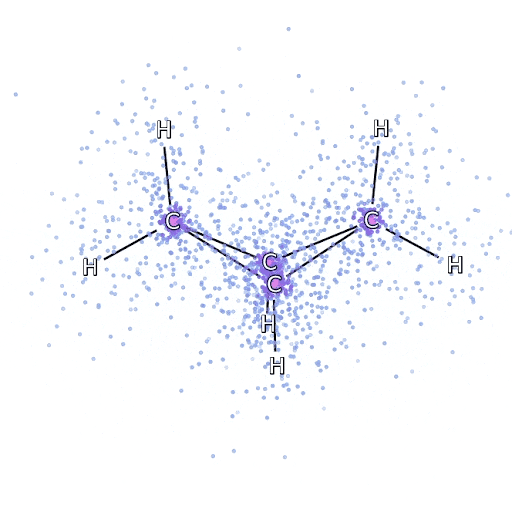
Este método funciona bien en moléculas en su configuración estable, pero se resiente cuando las uniones se tensan o se rompen, lo cual es crucial para entender las reacciones químicas. Aunque escala mejor que de manera exponencial, el método del racimo acoplado particular utilizamos escalas fijas como la octava potencia del número de electrones, por lo que solo puede aplicarse hasta cierto punto. Aplicamos FermiNet en moléculas progresivamente más grandes, comenzando con hidruro de litio y trabajando hacia el biciclobutano, el sistema más grande que hemos visto, con 30 electrones. En las moléculas pequeñas, FermiNet capturó un impresionante 99,8% de la diferencia entre la energía del racimo acoplado y la energía obtenida de un solo Slater. En el biciclobutano, FermiNet aún capturó al menos el 97% o más de esta energía de correlación, lo que es una gran lograda para tal sencilla aproximación. Representación gráfica de la fracción de energía de correlación que FermiNet captura en moléculas. De izquierda a derecha: hidruro de litio, nitrógeno, eteno, ozono, etanol y biciclobutano.